Alport Syndrome
Alport Syndrome is a rare inherited disease that can cause kidney failure, deafness and eye abnormalities (Alport UK). It can lead to the need for dialysis, and/or a kidney transplant, when people are in their 20s and 30s. The average age of commencing dialysis in X-linked Alports syndrome is about 35 years.
Even though rare, it is the second most common form of inherited kidney disease, after polycystic kidney disease (PKD).
Introduction – genetic basis
Alport syndrome is a form of ‘hereditary nephritis’ (kidney inflammation). It is a glomerular disease, i.e. it primarily damages the glomeruli of the kidneys.
This means it is a hereditary disorder caused by mutations in genes that lead to the production of collagen, specifically type IV. These mutations affect the structure and function of ‘basement membranes’ – including the kidney’s glomerular basement membrane (GBM) – in various tissues. This is what leads to the glomerular abnormalities in the diagrams below.
There is mutation (or absence) of the COL4A3, COL4A4, or COL4A5 genes.
What is collagen?
Collagen accounts for 30% of your body’s protein. Alport syndrome is caused by an abnormality in collagen in the body. Collagen is a bit like the cement of the body. It is part of the ‘building blocks’ that give parts of the body (e.g. skin, muscles and connective tissues) not supported by bone, their shape and strength.
Genetics of Alport syndrome (inheritance)
There are three genetic types of Alport syndrome. With all types of Alport syndrome, the kidneys are affected.
- X-linked Alport syndrome (XLAS). X-linked (related to the X chromosome) is the most common form of Alport Syndrome. About 80% of the people with this disease have the X-linked type. This means that it most commonly passes from a mother to a son(s). 50% of males require dialysis by age 25, and 90% develop ESRD by age 40. Females develop kidney failure less frequently and more slowly.
- Autosomal recessive Alport syndrome (ARAS). This is when both parents carry the abnormal gene and both parents pass the abnormal gene to the child. Both copies of the abnormal gene are needed to cause the autosomal recessive type of Alport Syndrome. 15% of all Alport cases are caused by this inheritance type. Both males and females with ARAS may develop kidney failure by age 20.
- Autosomal dominant Alport syndrome (ADAS). This happens when one parent has the disease and passes the abnormal gene to the child. In other words, only one copy of the abnormal gene is needed to cause the disease. 5% of all Alport cases are caused by this inheritance type. ADAS progresses slowly in both males and females, and kidney failure may not occur until later in life. Eye problems are uncommon in people with ADAS.
X-linked Alport syndrome in women
XLAS usually occurs in males but can occasionally happen in female carriers. Why? In cases of X-linked inheritance, the genetic defect causing the disease is on the X chromosome, a sex chromosome. Since men have only one copy of the X chromosome (they are XY), unlike women who have two (being XX), XLAS is more likely to affect men.
Hence women with one faulty copy of the X chromosome (female carriers) can develop the disease, but it is usually less severe in women because their other X chromosome can compensate.
However 95% of female XLAS carriers have haematuria by the time they reach adulthood. And the proportion of carriers developing significant renal disease in their lifetime is higher than previously thought, possibly as many as 25-30% reaching ESRD by 80 years. Most go undiagnosed or underdiagnosed due to variations in symptom severity and course of disease progression.
Kidney involvement
The most common and serious presentation of Alport syndrome is with chronic kidney disease (CKD), often with blood and protein in the urine (haematuria and proteinuria). Proteinuria can reach nephrotic levels (high) and this Alport syndrome is a cause of nephrotic syndrome.
CKD can progress and eventually lead to end-stage renal disease (ESRD), i.e. kidney failure – requiring dialysis and/or kidney transplantation. Hypertension (high blood pressure) is not usually an early feature.
Unfortunately, like many causes of CKD, Alport syndrome is often quite asymptomatic until its later stages. The best indicator of Alport syndrome (being a genetically inherited disease), is a being family member with Alport syndrome. If this is you, you need to be tested for it, even if you feel well.
Hearing loss
Sensorineural hearing loss is another common feature of Alport syndrome. This type of hearing loss affects the inner ear and can vary in severity. Hearing loss usually occurs before kidney failure.
This is because type IV collagen is an important component of inner ear structures, particularly the organ of Corti, that transform sound waves into nerve impulses for the brain. Altered versions of collagen IV in the inner ear impair its function, which can lead to hearing loss.
Eye abnormalities
Some individuals with Alport syndrome develop eye abnormalities, such as cataracts and lenticonus (abnormal bulging of the lens). This seldom leads to blindness.
This is because, in the eye, type IV collagen is important for maintaining the shape of the lens and the normal colour of the retina. Abnormal versions of collagen IV in the eye can result in misshapen lenses and an abnormally coloured retina.
Diagnosis
Diagnosis is typically made through a combination of medical assessment by a hospital kidney specialist (nephrologist), focusing on family history; blood and urine tests; and in some, renal (kidney) biopsy, and genetic testing.
But Alport’s syndrome can be diagnosed in different ways. Firstly, if Alport’s syndrome is known to run in the family, a simple blood test for CKD and urine test for blood and protein may be enough, to be fairly sure of the diagnosis.
Secondly, for some with CKD, where there is uncertainty, a kidney biopsy (sample of kidney removed with a needle) may be necessary. Alport’s syndrome has a particular appearance in the kidney when examined under the microscope. Lastly, testing for the abnormal gene in Alport’s syndrome is sometimes needed.
How does it affect the glomerulus?


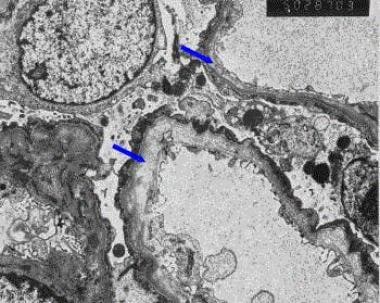
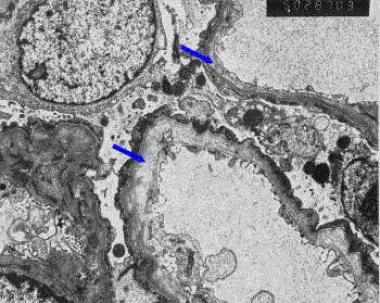
Alport Syndrome abnormalities on a renal biopsy (using an electron microscope, EM). EM findings are needed to diagnose it for certain. With normal (light) microscopy, the findings are non-specific (i.e. can occur with other kidney diseases).
Management and treatment
There is no cure for Alport syndrome, but treatment focuses on slowing the progression of kidney disease. This involves tight blood pressure control; especially using angiotensin-converting enzyme (ACE) inhibitors (e.g. Ramipril) and angiotensin receptor blockers (ARBs, e.g. Losartan). These can slow down the progression of kidney disease and improve outcomes.
A newer group of drugs (originally for diabetes) called sodium-glucose cotransporter 2 (SGLT-2) inhibitors (such as Dapagliflozin) may also be useful in addition to ACE/ARBs.
Nonetheless, despite treatment, dialysis and kidney transplantation may be eventually required. Kidney transplants are often very successful in people with Alport syndrome.
Note. A minority of patients with Alport syndrome develop anti-GBM disease in their new kidney after renal transplantation.
Genetic counselling
Genetic counselling is essential for individuals with Alport syndrome or those at risk of carrying the genetic mutation. It can help in making informed family planning decisions.
It’s important to note that Alport syndrome can manifest differently in different individuals, and the severity of its features can vary.
Why is it called Alport Syndrome?
Alport’s syndrome is named after Dr A. Cecil Alport, who recognised families with the syndrome in 1927. He was born in 1880 in South Africa, and graduated as a doctor in Edinburgh. At first he worked in Johannesburg (owning a small gold mine). After the First World War he moved to St Mary’s Hospital, Paddington, London, where he was working when he described the syndrome of hereditary renal failure and deafness. Later he worked in Cairo, and died in 1959.
Other names for Alports syndrome
Congenital hereditary haematuria
Haematuria-nephropathy-deafness syndrome
Haematuric hereditary nephritis
Haemorrhagic familial nephritis
Haemorrhagic hereditary nephritis
Hereditary familial congenital haemorrhagic nephritis
Hereditary haematuria syndrome
Hereditary interstitial pyelonephritis
Hereditary nephritis
Summary
We have described what is Alport Syndrome. We hope it has been helpful.
Other resources
These are review articles or guidelines for health professionals: Savige, 2013, Watson, 2023
These are the latest research articles on pubmed.
These are other sources of information.
Alport research hub
Alport syndrome foundation (US charity)
Alport UK (UK charity) – patient information contributed to this article
Cleveland clinic
Kidney care UK (UK charity)
Kidney research UK
Medline plus
National kidney federation (UK charity)
National kidney foundation (US charity)
Penn medicine
UCLA health
UK kidney association
There is an overlap between Alport Syndrome and Thin Membrane Disease.
References
- Zhang Y, Ding J. Renal, auricular, and ocular outcomes of Alport syndrome and their current management. Pediatr Nephrol. 2018 Aug;33(8):1309-1316. [PubMed]
- Watson S, Padala SA, Hashmi MF, Bush JS. StatPearls [Internet]. StatPearls Publishing; Treasure Island (FL): Feb 19, 2023. Alport Syndrome. [PubMed]
- Kashtan C. Alport syndrome: facts and opinions. F1000Res. 2017;6:50. [PMC free article] [PubMed]
- Savige J. Alport syndrome: deducing the mode of inheritance from the presence of haematuria in family members. Pediatr Nephrol. 2020 Jan;35(1):59-66. [PubMed]
- Kashtan CE. Renal transplantation in patients with Alport syndrome: patient selection, outcomes, and donor evaluation. Int J Nephrol Renovasc Dis. 2018;11:267-270. [PMC free article] [PubMed]
- Vos P, Zietse R, van Geel M, Brooks AS, Cransberg K. Diagnosing Alport Syndrome: Lessons from the Pediatric Ward. Nephron. 2018;140(3):203-210. [PubMed]
- Katsuma A, Nakada Y, Yamamoto I, Horita S, Furusawa M, Unagami K, Katsumata H, Okumi M, Ishida H, Yokoo T, Tanabe K., Japan Academic Consortium of Kidney Transplantation (JACK). Long-term survival in Japanese renal transplant recipients with Alport syndrome: a retrospective study. BMC Nephrol. 2018 Oct 03;19(1):249. [PMC free article] [PubMed]
- Crockett DK, Pont-Kingdon G, Gedge F, Sumner K, Seamons R, Lyon E. The Alport syndrome COL4A5 variant database. Hum Mutat. 2010 Aug;31(8):E1652-7. [PubMed]
- Hashimura Y, Nozu K, Kaito H, Nakanishi K, Fu XJ, Ohtsubo H, Hashimoto F, Oka M, Ninchoji T, Ishimori S, Morisada N, Matsunoshita N, Kamiyoshi N, Yoshikawa N, Iijima K. Milder clinical aspects of X-linked Alport syndrome in men positive for the collagen IV α5 chain. Kidney Int. 2014 May;85(5):1208-13. [PubMed]
- Nozu K, Minamikawa S, Yamada S, Oka M, Yanagita M, Morisada N, Fujinaga S, Nagano C, Gotoh Y, Takahashi E, Morishita T, Yamamura T, Ninchoji T, Kaito H, Morioka I, Nakanishi K, Vorechovsky I, Iijima K. Characterization of contiguous gene deletions in COL4A6 and COL4A5 in Alport syndrome-diffuse leiomyomatosis. J Hum Genet. 2017 Jul;62(7):733-735. [PubMed]
- Hicks J, Mierau G, Wartchow E, Eldin K. Renal diseases associated with hematuria in children and adolescents: a brief tutorial. Ultrastruct Pathol. 2012 Feb;36(1):1-18. [PubMed]
- Warady BA, Agarwal R, Bangalore S, Chapman A, Levin A, Stenvinkel P, Toto RD, Chertow GM. Alport Syndrome Classification and Management. Kidney Med. 2020 Sep-Oct;2(5):639-649. [PMC free article] [PubMed]
- Savige J, Colville D, Rheault M, Gear S, Lennon R, Lagas S, Finlay M, Flinter F. Alport Syndrome in Women and Girls. Clin J Am Soc Nephrol. 2016 Sep 07;11(9):1713-1720. [PMC free article] [PubMed]
- Deltas C, Pierides A, Voskarides K. Molecular genetics of familial hematuric diseases. Nephrol Dial Transplant. 2013 Dec;28(12):2946-60. [PubMed]
- Fallerini C, Dosa L, Tita R, Del Prete D, Feriozzi S, Gai G, Clementi M, La Manna A, Miglietti N, Mancini R, Mandrile G, Ghiggeri GM, Piaggio G, Brancati F, Diano L, Frate E, Pinciaroli AR, Giani M, Castorina P, Bresin E, Giachino D, De Marchi M, Mari F, Bruttini M, Renieri A, Ariani F. Unbiased next generation sequencing analysis confirms the existence of autosomal dominant Alport syndrome in a relevant fraction of cases. Clin Genet. 2014 Sep;86(3):252-7. [PubMed]
- Morinière V, Dahan K, Hilbert P, Lison M, Lebbah S, Topa A, Bole-Feysot C, Pruvost S, Nitschke P, Plaisier E, Knebelmann B, Macher MA, Noel LH, Gubler MC, Antignac C, Heidet L. Improving mutation screening in familial hematuric nephropathies through next generation sequencing. J Am Soc Nephrol. 2014 Dec;25(12):2740-51. [PMC free article] [PubMed]
- Delimont D, Dufek BM, Meehan DT, Zallocchi M, Gratton MA, Phillips G, Cosgrove D. Laminin α2-mediated focal adhesion kinase activation triggers Alport glomerular pathogenesis. PLoS One. 2014;9(6):e99083. [PMC free article] [PubMed]
- Dufek B, Meehan DT, Delimont D, Cheung L, Gratton MA, Phillips G, Song W, Liu S, Cosgrove D. Endothelin A receptor activation on mesangial cells initiates Alport glomerular disease. Kidney Int. 2016 Aug;90(2):300-310. [PMC free article] [PubMed]
- Gunwar S, Ballester F, Noelken ME, Sado Y, Ninomiya Y, Hudson BG. Glomerular basement membrane. Identification of a novel disulfide-cross-linked network of alpha3, alpha4, and alpha5 chains of type IV collagen and its implications for the pathogenesis of Alport syndrome. J Biol Chem. 1998 Apr 10;273(15):8767-75. [PubMed]
- Cosgrove D, Rodgers K, Meehan D, Miller C, Bovard K, Gilroy A, Gardner H, Kotelianski V, Gotwals P, Amatucci A, Kalluri R. Integrin alpha1beta1 and transforming growth factor-beta1 play distinct roles in alport glomerular pathogenesis and serve as dual targets for metabolic therapy. Am J Pathol. 2000 Nov;157(5):1649-59. [PMC free article] [PubMed]
- Cosgrove D. Glomerular pathology in Alport syndrome: a molecular perspective. Pediatr Nephrol. 2012 Jun;27(6):885-90. [PMC free article] [PubMed]
- Olaru F, Luo W, Wang XP, Ge L, Hertz JM, Kashtan CE, Sado Y, Segal Y, Hudson BG, Borza DB. Quaternary epitopes of α345(IV) collagen initiate Alport post-transplant anti-GBM nephritis. J Am Soc Nephrol. 2013 May;24(6):889-95. [PMC free article] [PubMed]
- Wang XP, Fogo AB, Colon S, Giannico G, Abul-Ezz SR, Miner JH, Borza DB. Distinct epitopes for anti-glomerular basement membrane alport alloantibodies and goodpasture autoantibodies within the noncollagenous domain of alpha3(IV) collagen: a janus-faced antigen. J Am Soc Nephrol. 2005 Dec;16(12):3563-71. [PubMed]
- Borza DB. Autoepitopes and alloepitopes of type IV collagen: role in the molecular pathogenesis of anti-GBM antibody glomerulonephritis. Nephron Exp Nephrol. 2007;106(2):e37-43. [PubMed]
- Kashtan CE. Alport syndrome and thin glomerular basement membrane disease. J Am Soc Nephrol. 1998 Sep;9(9):1736-50. [PubMed]
- Thorner PS. Alport syndrome and thin basement membrane nephropathy. Nephron Clin Pract. 2007;106(2):c82-8. [PubMed]
- Patey-Mariaud de Serre N, Garfa M, Bessiéres B, Noël LH, Knebelmann B. Collagen alpha5 and alpha2(IV) chain coexpression: analysis of skin biopsies of Alport patients. Kidney Int. 2007 Aug;72(4):512-6. [PubMed]
- Gubler MC. Diagnosis of Alport syndrome without biopsy? Pediatr Nephrol. 2007 May;22(5):621-5. [PubMed]
- Gubler M, Levy M, Broyer M, Naizot C, Gonzales G, Perrin D, Habib R. Alport’s syndrome. A report of 58 cases and a review of the literature. Am J Med. 1981 Mar;70(3):493-505. [PubMed]
- Izzedine H, Tankere F, Launay-Vacher V, Deray G. Ear and kidney syndromes: molecular versus clinical approach. Kidney Int. 2004 Feb;65(2):369-85. [PubMed].
Last Reviewed on 14 June 2024