Renal thrombotic microangiopathy (TMA)
This article focuses on both thrombotic microangiopathy (TMA) in general and renal causes in particular.
Thrombotic microangiopathy (TMA) is a diverse group of conditions that classically present with microangiopathic haemolytic anaemia (MAHA) and thrombocytopaenia together with organ dysfunction (including the kidneys in some cases).
MAHA is a subgroup of haemolytic anaemias (loss of red blood cells through destruction) caused by factors damaging the endothelium (lining of small arteriolar blood vessels) all over the body.
That damage leads to organ dysfunction (e.g. kidney, liver or brain) which can present in very different ways. It is also the underlying mechanism of eclampsia which can occur in the third trimester of pregnancy.
TMAs are rare. But there can be devastating consequences to missing the diagnosis. They are identified by the finding of anaemia with fragmented red blood cells (see below) on looking at a sample of blood under a microscope.
There are many causes. Renal causes (often called haemolytic uraemic syndrome, HUS) are characterised by the ‘classic TMA/MAHA triad’ of:
- Haemolytic anaemia (reduced haemoglobin, with haemolysis present on blood film)
- Thrombocytopenia (low platelets)
- Acute kidney injury (AKI) – usually with signs of fluid overload.
In TMAs, the endothelial layer of small vessels is damaged with resulting fibrin deposition and platelet aggregation (sticking together). As red blood cells travel through these damaged vessels, they are fragmented resulting in intravascular haemolysis.
So, let’s discuss renal thrombotic microangiopathy (TMA) more. But we need a bit more background first.
How do TMAs damage tissue?
Whichever organ is involved the mechanism of damage is the same, i.e. clumps of fragmented RBCs and platelets stick on to the damaged endothelium blocking them (thrombosis).
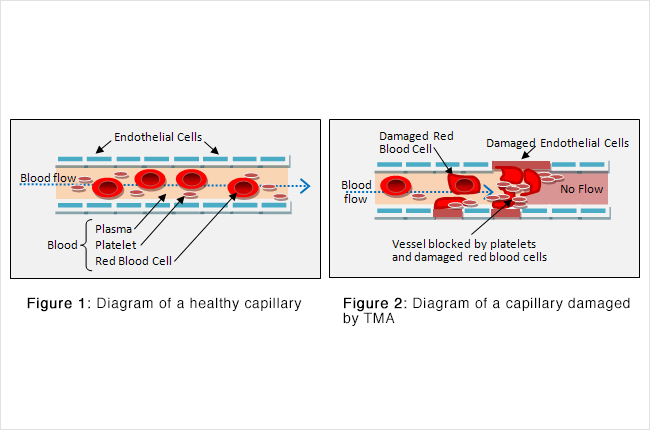
The red cell fragments are also increasingly targeted for destruction in the spleen, due to their narrow passage through obstructed vessel lumina.
Why is it called ‘thrombotic microangiopathy’?
- ‘Microangiopathy’ means a ‘small blood vessel problem’
- ‘Thrombotic’ means that blood clots are involved.
We now describe thrombotic microangiopathy, with a focus on renal causes.
Clinical features
These are dominated by the underlying cause. In this way the clinical features may be very different.
In non-renal causes, the third part (AKI) of the classic triad may be replaced with (or in addition to) the underlying cause, e.g.
- Liver impairment in HELLP
- Severe hypertension (with target organ damage) and/or being unconscious in accelerated hypertension
- Neurological features in TTP (including confusion and fits)
- Proteinuria (and/or being pregnant) in eclampsia.
The low platelets may show itself as a thrombocytopenic (TCP) rash, called petechia and purpura. This can be subtle.
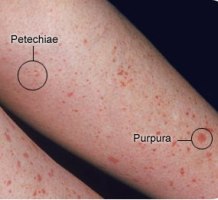
Thrombocytopenic rash
Causes
- Classical (typical) haemolytic uraemic syndrome (HUS) – often secondary to infection or drugs
- Atypical haemolytic uraemic syndrome – due to an autoimmune or inherited causes (see below)
- Thrombotic thrombocytopenic purpura (TTP) – more common in children. Also due to typical and atypical forms (see below)
- Disseminated intravascular coagulation (DIC) – usually due to sepsis
- HELLP syndrome (liver)
- Accelerated hypertension (hypertensive emergency) – i.e. severe hypertension with target-organ damage
- Scleroderma renal crisis (SRC)
- Mechanical cardiac valves (or ‘Waring Blender syndrome’)
- Kasabach–Merritt syndrome (Osman, 2013)
- Infections (e.g. influenza A, bacterial endocarditis, HIV, parvovirus B19, EBV, CMV, hepatitis viruses, COVID-19 (Vrecko, 2022), malaria)
- Drugs (e.g. ciclosporin/tacrolimus, quinine, ticlopidine, clopidogrel, IFN, VEGF inhibitors)
- Autoimmune disease (vasculitides such as polyarteritis nodosa (PAN), granulomatosis with polyangiitis, and antiphospholipid antibody syndrome (APAS), and systemic lupus erythematosus, (SLE) (Kello, 2019))
- Glomerular disorders – membranous nephropathy, IgA nephropathy, C3 nephropathy (Chabannes, 2023), FSGS, ANCA-positive vasculitis, mesangiocapillary glomerulonephritis
- Metabolic – cobalamin C deficiency due to MMACHC gene mutations
- Malignancy – systemic cancers or secondary to chemotherapy
- Others diseases: eclampsia (Fakhouri, 2020), kidney transplant rejection (Epperla, 2020), paroxysmal nocturnal haemoglobinuria (PNH), toxins (including snake bite; Rahmani, 2018).
Typical vs atypical HUS
The most common form of HUS (typical HUS) often follows a diarrhoeal illness caused by O157 (shiga toxin)–producing Escherichia coli. They can be mini-outbreaks in children who have visited animal petting farms (e.g. after school trips). Other bacteria cause the syndrome as well.
Atypical HUS is caused by a severe deficiency of the enzyme ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) due to acquired autoantibodies or, rarely, genetic mutations (associated with complement-mediated damage). In this way, the same rare disease can be seen on one family.
ADAMTS13 is a normal enzyme in plasma that cleaves large forms of the coagulation protein von Willebrand factor into smaller functional subunits. The acquired form of ADAMT13 associated- HUS (and TTP) is thought to be due to an autoantibody against ADAMTS13, and the inherited form is due to a genetic deficiency of ADAMTS13.
Diagnosis
This is based on clinical suspicion, clinical features and (often of underlying cause) and investigations:
- FBC (full blood count) showing haemolysis with fragmented red blood cells (schistocytes) and low platelets. So TMAs may be suspected based on routine medical laboratory tests such as FBC. Automated analysers (the machines that perform routine FBCs in most hospitals) are designed to flag: (1) blood specimens that contain abnormal amounts of red blood cell fragments; and (2) low platelets
- Other investigations (e.g. AKI on U+E, abnormal liver enzymes in HELLP)
- Renal biopsy (occasionally).
If such changes are linked to – for example AKI (in HUS) or abnormal liver enzymes (HELLP) – the combination is a classical ‘clinical/laboratory picture’, which is obvious to experienced specialist senior health professionals (e.g. nephrologists, liver physicians, haematologists and obstetricians).
TMAs are rare so can easily be missed by junior health professionals. So if a junior thinks of one, and they are not sure, they would discuss the situation with a senior ASAP (yes, that means 2am as well!).
Why? Sometimes emergency action is required, e.g, delivering the baby now in eclampsia, and/or IV anti-hypertensive agents, broad spectrum antibiotics in DIC/sepsis, plasma exchange in aHUS.
Samples for ADAMTS13 should be sent to a specialist laboratory. This may take a week or more to come back.

Typical findings in a kidney biopsy of a patient with TMA. They are often not done due to the low platelet count and/or high blood pressure.
Note. There is not always a ‘full house’ of laboratory features initially. For example in a renal TMA (HUS), there can be AKI and haemolytic anaemia but normal (or low-normal) platelets initially. This is why a high degree of suspicion is necessary.
Pathophysiology
In all causes, the mechanism of TMA is the formation of a fibrin mesh due to increased activation of the coagulation system. Fibrin is a protein formed from fibrinogen during the clotting of blood.
The red blood cells are physically cut by these protein networks. The resulting fragments are the schistocytes observed under a light microscope (see blood film below).

Peripheral blood film showing classic findings of thrombotic microangiopathy. The image shows features of thrombocytopenia and microangiopathic haemolysis. Arrows show red blood cell fragments (i.e. schistocytes).
Treatment
The primary treatment is to remove (or treat) the underlying cause, e.g:
- Delivering the baby (now!) in eclampsia
- IV anti-hypertensive agents in accelerated hypertension
- Broad spectrum antibiotics in DIC/sepsis.
For patients with typical HUS (e.g. Shiga toxin-producing E. coli), treatment involves supportive care (e.g. fluid rehydration, red cell transfusions). Antimicrobial treatment may worsen the disease, depending on the bacterial strain and antibiotic class. Plasma exchange is rarely needed.
For patients with atypical HUS, plasma exchange should be administered initially, and soon. Corticosteroids (e.g. IV methylprednisolone or oral prednisolone) are frequently added to plasma exchange as part of initial therapy.
Also, eculizumab (Zafar, 2023), a mono-clonal antibody against the terminal complement component C5, should be considered. This usually involves discussion with a regional HUS centre (e.g. ones that carry out the ADAMTS13 assay).
Kidney specialist doctors (nephrologists) should avoid treatment delays and misdiagnoses by not waiting to base treatment decisions on results of ADAMTS13 testing.
Platelets and cryoprecipitate are contraindicated as they facilitate further clot formation and breaking down of RBCs.
What is the treatment for refractory or relapsed atypical HUS?
Atypical HUS may be refractory (not responding well to initial treatment) or relapse. Refractory aHUS is generally defined as the lack of rise in platelet count, persistent haemolysis, lack of symptom resolution or new clinical signs (e.g. neurological) after five to seven days of plasma exchange.
Relapse is defined as disease recurrence after remission, usually 30 days after the last plasma exchange treatment.
Relapsed or refractory aHUS can be managed successfully by increasing the intensity or frequency of plasma exchanges, and adding or increasing doses of immunosuppressant therapy, including steroids.
Rituximab has been also been shown to be effective in this situation, and some investigators have used it as initial therapy. The proteasome inhibitor bortezomib has also been effective for some patients, although controlled trials have not been done.
Other treatments include ciclosporin (strangely, as it can also cause the disease), cyclophosphamide, vincristine, N-acetylcysteine, and splenectomy.
Emerging therapies include caplacizumab, a novel anti–von Willebrand factor nanobody, and recombinant ADAMTS13.
For patients with relapsed or refractory atypical HUS who require renal transplantation, pre-emptive use of eculizumab can improve transplantation outcomes. Also combined liver–kidney transplantation can restore renal function and increase the levels of complement regulatory proteins that are produced by the liver. Patients with refractory atypical HUS should be re-evaluated to ensure that secondary causes are not missed.
Again, all such treatments should be discussed with a regional HUS centre.
Summary
We have described renal thrombotic microangiopathy (TMA), focusing on aHUS. We hope it has been helpful.
Other resources
These are review articles: Arnold, 2017, Htet, 2020 and Thompson, 2022.
The appearances under the microscope are shown by Lusco, 2016.
Last Reviewed on 28 January 2024